

The European Union aims at entering a new era of clinical trials by enforcing the steps towards personalized medicine. Instead of searching for the best therapy to treat a certain diagnosis, they are moving towards pursuing the best suitable individual therapy. To meet this approach, Big Data technologies have developed new therapies and potential positive effects by using data bases. Aggregated and integrated Data bases, more precisely the raw data, are the new important raw material (such as oil was in the times of industrialization) with high potential effects for future innovations, especially within the pharmaceutical industry. The aggregated data sets function as knowledge resources for the pharma sector.
What is the legal reasoning to obligatory publish clinical trial data?
The development of new drugs is the financial basis for the pharmaceutical industry worldwide. In the European Union, the pharmaceutical market is regulated by the European Medicine Agency (EMA), as the European Unions’ authorization agency. Hereby, the EMA functions as a guardian to adequately balance relevant European fundamental rights:
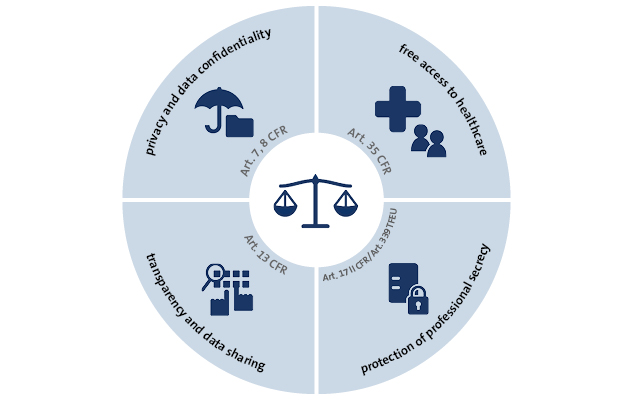
The EMA has found the ideal balance of the contradicting interests by declaring clinical trial data as non-confidential commercial information, regardless of the fact that data obtained through clinical trials is a main innovative resource for the pharmaceutical industry. The EMA intended to balance the interests of the pharma industry against the protection of public health. Following the argumentation of the EMA, it decided to empower European medical research and improve the use of existing products by the obligatory publication of clinical trial data with only a few exceptions.[1] The European regulation 536/2014 and further transparency policies main goal was to establish an European database accessed through an EU portal as a central electronic communication platform[2]. The database must contain relevant information, the clinical trials’ summary, the protocols and the clinical study reports of conducted clinical trials, in order to obtain the registration and enable market access.[3] It’s important to emphasize the obligatory publication of linking data to the drug[4], meaning that, even information on clinical trials conducted outside of the European Unions’ territory, should be included within the drug registration request.
What about the GDPR?
In the future, the General Data Protection Framework generally increases the data protection standards to ensure a high level of data protection and privacy. This contrasts directly with the focus of having a comprehensive data publication by the EMA. It is unclear whether the contradictory balance of these points of view might be influenced towards disrupting the equilibrium between these interests’ due to the rising importance of data privacy.
Either way and luckily for the EMA, aggregated data does fall in the scope of the GDPR, if it is impossible to trace the information to the data subjects.[5] Whether it is always possible to anonymize the personal data in question depends on diverse factors, such as the drug’s scope of application. A problem concludes the risk in small studies considering rare diseases: Data mining and the connection to data portals enables a possible reidentification of the individual patient.[6] Hereby, the aggregated data might be deduced to personal data and the scope of application of the GDPR might be broadened.
EMA Transparency Roadmap
The EMA transparency policies consist of two phases of the obligatory publication. The first phase includes the publication of all study reports no matter whether the drug authorization is requested as a central, decentral or fully EU national procedure. The data published is pseudonymized but not anonymized. The second phase involves the proactive publication of all anonymized individual patient data, “raw data”. Raw data are the most valuable part of the drug approval documents. In order to exclude the scope of the GDPR any potential option to reidentify patients on the bases of raw data has to be excluded to enter the second phase of the enforced data publication.
In September 2016, the first dossiers were published by the EMA on a preliminary database on their web page. Considering the self-information given by the EMA on their web page from October 2016, the publication on the EMA website will be mandatory in future. This refers to the applications submitted by pharmaceutical companies under the centralized procedure.[7] The POLICY/0070[8] exempts commercially confidential information, information of acting people and identifiable aspects of study participants from the obligatory publication. The pharmaceutical companies can exclude single words, numbers or text parts[9] from being published, if the potential advantages for competitors rise from obtaining this information[10], but the EMA has to approve the edition. This implies a power to minimize the information to be restrained from the public.
Furthermore, the EMA terms of use worldwide limit every use of data for general information and non-commercial purposes to inhibit unfair commercial use.[11] Further effects might and will be recognized in 2018, when the EU portal and EU database are planned to come into force.
Situation today
Many questions and doubts still remain considering the future influence of the GDPR and the potential risks of individual health data. Due to insecure events such as the real importance of the EMA database website any comments regarding potential influence or risks of exposing the data to be published in the different phases of the EMA policies remain uncertain.
We will update you on further changes in regard to the obligatory sharing of clinical data by the EMA.
[1] See p. 3, 11,
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/10/
WC500195084.pdf
[2] Recital 66 Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products
for human use, repealing Directive 2001/29/EC, OJ L 158/1 (27.5.2014).
[3] Recital 67 Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products
for human use, repealing Directive 2001/29/EC, OJ L 158/1 (27.5.2014); See p. 28,
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2015/10/
WC500195084.pd>(last visited 22 April 2017.
[4] Recital 67 Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products
for human use, repealing Directive 2001/29/EC, OJ L 158/1 (27.5.2014).
[5] Art. 89 Sec. 2 GDPR.
[6] POLICY 0070 of 2014 European Medicines Agency policy on publication of clinical data for medicinal products for human use, EMA/240810/2013 (2 October 2014).
[7] http://www.ema.europa.eu/ema/?curl=pages/special_topics/general/general
[8] POLICY 0070 of 2014 European Medicines Agency policy on publication of clinical data for medicinal products for human use, EMA/240810/2013 (2 October 2014).
[9] External guidance of 2016 on the implementation of the European Medicines Agency policy on the publication of clinical data for medicinal products for human use, EMA/90915/2016p. 47.
[10] External guidance of 2016 on the implementation of the European Medicines Agency policy on the publication of clinical data for medicinal products for human use, EMA/90915/2016, p. 46ff.
[11] Annex 1f. POLICY 0070 of 2014 European Medicines Agency policy on publication of clinical data for medicinal products for human use, EMA/240810/2013 (2 October 2014).